Exercises for today¶
You will be doing 3 exercises to become familar with some of the bioinformatic tools that are used by researchers today. These tools are currently being used actively by researchers who are studying microbial genomes.
1. sra-tools¶
The first is sra-tools, which are a suite of tools that you can use to download data from NCBI sequence read archives (SRA) (see here: https://www.ncbi.nlm.nih.gov/sra). This database is essential to a lot of researchers. This is where they will deposit raw sequences that came out of sequencing instruments. You can search for research project focusing on a certain topic and download the data they have deposited to SRA. It is publicly accessible.
For example, if you type “Yellowstone hot spring” in the search box, you will see a list of accessions with links to the data. An example is here:
https://www.ncbi.nlm.nih.gov/sra/SRX9071323
If you click on that link, you will see that the study is titled “Seasonal hydrologic and geologic forcing drive hot spring geochemistry and microbial biodiversity”. And there is a link below that says “Run, # of spots, # of Bases” etc. To download this sequence data, you will need to click on the link that starts with “SRR”, click on the “Data access” tab, then click on the link provided next to the “run”.
So it becomes very cumbersome and difficult to navigate these pages to access and download the data. This is where the sra-tools becomes very useful. You can download the raw sequence reads deposited to SRA by just typing the following command with the accession number next to the [accn].
fasterq-dump --split-3 SRR12584454
The “SRR12584454” is the actual run accession number for that particular study. This is actually the number you need to enter when trying to download these raw sequences from NCBI SRA.
First, you might want to explore what fasterq-dump means by typing fasterq-dump -h. -h just means you are trying to bring up help menu to list the options available with this command.
[1]:
%%bash
fasterq-dump -h
Usage:
fasterq-dump <path> [options]
Options:
-o|--outfile output-file
-O|--outdir output-dir
-b|--bufsize size of file-buffer dflt=1MB
-c|--curcache size of cursor-cache dflt=10MB
-m|--mem memory limit for sorting dflt=100MB
-t|--temp where to put temp. files dflt=curr dir
-e|--threads how many thread dflt=6
-p|--progress show progress
-x|--details print details
-s|--split-spot split spots into reads
-S|--split-files write reads into different files
-3|--split-3 writes single reads in special file
--concatenate-reads writes whole spots into one file
-Z|--stdout print output to stdout
-f|--force force to overwrite existing file(s)
-N|--rowid-as-name use row-id as name
--skip-technical skip technical reads
--include-technical include technical reads
-P|--print-read-nr print read-numbers
-M|--min-read-len filter by sequence-len
--table which seq-table to use in case of pacbio
--strict terminate on invalid read
-B|--bases filter by bases
-A|--append append to output-file
-h|--help Output brief explanation for the program.
-V|--version Display the version of the program then
quit.
-L|--log-level <level> Logging level as number or enum string. One
of (fatal|sys|int|err|warn|info|debug) or
(0-6) Current/default is warn
-v|--verbose Increase the verbosity of the program
status messages. Use multiple times for more
verbosity. Negates quiet.
-q|--quiet Turn off all status messages for the
program. Negated by verbose.
--option-file <file> Read more options and parameters from the
file.
/Users/jsaw/miniconda3/bin/fasterq-dump : 2.10.1
As you can see, there are multiple options available with this tool and you can even specify things like memory limitation for this command to run and the number of threads you want to use with this tool. Before actually typing the command, I want you to make sure that you change your directory to “exercises” directory that I made you create last week. I am giving you an example of where I created the folders and organize them.
[4]:
%%bash
cd ~/workspace/
tree MicrobialGenomicsLab
MicrobialGenomicsLab
├── data
├── exercises
├── repositories
└── tools
4 directories, 0 files
Here, you will see that I created the 4 folders inside “MicrobialGenomicsLab” folder which resides under “workspace”. Navigate into the exercises folder and run the fasterq-dump tool with the example command that I wrote earlier.
fasterq-dump --split-3 SRR12584454
The command will start downloading the raw sequence files from SRA to your “exercises” folder. It may take a few minutes and you will see the report printed to the screen once it’s done. Usually it will not tell you what is going on but you can increase the verbosity of the screen output by typing like this:
fasterq-dump -v --split-3 SRR12584454
Once it’s done, you can type ls -la to see what files the tool produced.
[5]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
ls -la
total 165888
drwxr-xr-x 4 jsaw 982768932 128 Sep 9 09:05 .
drwxr-xr-x 6 jsaw 982768932 192 Sep 9 08:58 ..
-rw-r--r-- 1 jsaw 982768932 70168140 Sep 9 09:05 SRR12584454_1.fastq
-rw-r--r-- 1 jsaw 982768932 70168140 Sep 9 09:05 SRR12584454_2.fastq
Now, you should see 2 files being produced by the fasterq-dump tool. Both files end with a “.fastq” file extension. These are fastq-formatted files that can be observed/analyzed with tools like fastqc or bbmap. Now, try to see what the contents of these fastq files look like.
[9]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
head -8 SRR12584454_1.fastq
@SRR12584454.1 1 length=301
GTGCCAGCCGCCGCGGTAATACCAGCCCCGCGAGTGGTCGGGACTCTTACTGGGCCTAAAGCGCCCGTAGCCGGCCCGACAAGTCACTCCTTAAAGACCCCGGCTCAACCGGGGGAATGGGGGTGATACTGTCGGGCTAGGGGGCGGAAGAGGCCAGCGGTACTCCCGGAGTAGGGGCGAAATCCTCAGATCCCGGGAGGACCACCAGTGGCGAAAGCGGCTGGCTAGAACGCGCCCGACGGTGGGGGGCGAAAGGCGGGGCAGAGAAAGGGATTAGAAAACCCTTGAGGTCAGATGGGAA
+SRR12584454.1 1 length=301
CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG@FCFGGGGGGGGGGGGGGGGGGGAFGGGGGGGEGGCCGGGGGCCGGGGGGGGGECGCFFGGGGGGGEFGGGGGGGGC6:CGGCGEEGGGGGGGGGG=:8EEC6C:C:?C*2<:<A<959*:763**:,;)/:EG(1):*-(2/><C@)-((.<F483((2))0:9,;855*.-*((,)(--)+20+
@SRR12584454.2 2 length=301
GTGTCAGCAGCCGCGGTAATACGGAGGGTGCGAGCGTTACTCGGAATTACTGGGCGTAAAGCGCGCGTAGGTGGTTTGTTAAGTTGGATGTGAAATCCCCGGGCTCAACCTGGGAACTGCATTCCAAACTGACGAGCTAGAGTATGGTAGAGGGTGGTGGAATTTCCTGTGTAGCGGTGAAATGCGTAGATATAGGAAGGAACACCAGTGGCGAAGGCGACCACCTGGAATGATACTGACAGTGAGGTGCGAAAGCGGGGGGAGCAAACAGGATTAGATACCCCGGTAGTCCAGATCGGAA
+SRR12584454.2 2 length=301
CCCCCGGGAFGGGGGGGGGGGG,6FGG788C+CFGGGG7,9EF,+6CFGGGGFGGGGGGGGGG7+@FGGG,:EDGG844BFGG<,F,,CFG9FFGCGGG+++@FGGGGG3F,BF<F+=FFGGGG,,@FGGG,3>++@F3DFGG,=3FG9,@FG:3CF>FGGGGG,FG,?F<FFFGGGGGGGGGGGGGGGGGGGCFG,BFGGGCFGGFGGGFEGGGGG=G*?E8*2CFG6<+97CCEGD?E9<<7>CGC5:D3C>1:/3>DDF@118?C*7;C?DFB<C*@7CC)014(/8)294<>?EAE5
I typed head -8 SRR12584454_1.fastq to print the first 8 lines of the fastq file to the terminal screen. As you can see, the file stores a string of letters representing the bases of DNA and other weird characters. Try to see if you can see a pattern here. Each record for a single fragment of DNA is represented by 4 lines.
The first line starts with a ‘@’ followed by a string of alphabets and numbers. This should be unique to each sequence record. The actual DNA sequence is on the 2nd line. The third line starts with ‘+’ and same identifier as the first line. The 4th line contains characters that represent sequence quality for each of the DNA bases shown on the 2nd line. This is very important information for tools like fastqc or bbmap that relies on this information to assess sequence quality.
To understand more about how fastq files are encoded, see here: https://en.wikipedia.org/wiki/FASTQ_format
You see 2 fastq files that end with “_1” and “_2” in their file names. The reason for that is Illumina sequences that produced these files is usually run to sequence two ends of a single DNA fragment and for each DNA fragment that was sequenced, you have two sequences that you know are physically linked and therefore we call them “read pairs”. Pairing information is very useful for genome assembly and mapping. We will come to that in later labs.
Now that you have 2 fastq files, what do we do with them? First, what would you do if you want to know how many sequences are in these files? You can type something like this:
grep -c "@SRR12584454" SRR12584454_1.fastq
grep -c "@SRR12584454" SRR12584454_2.fastq
And both should return the same numbers. I will show you the example below. These are relatively small files by Illumina sequencing standards (only about 67 Mbp) so it is ok to run this grep command. I would not recommend this with files larger than several gigabytes large.
[10]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
grep -c "@SRR12584454" SRR12584454_1.fastq
grep -c "@SRR12584454" SRR12584454_2.fastq
103842
103842
As you can see, I am searching for the pattern “@SRR12584454” in each fastq file because I know that this identifier is present in each of the header line of the sequences. grep -c just counts the occurrences of these identifiers instead of printing the matches to screen. I see that both fastq files contain 103842 sequences. This is very small number. Usually you will have millions of sequences per fastq file. So there is no way for you to manually inspect each and every one of these
sequences for their quality. This is where the FastQC tool comes in.
2. FastQC¶
FastQC tool can be found here: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ but also through conda and you should have installed it through conda by now.
It is a tool to analyze high-throughput sequencing data such as those produced by Illumina sequencing technology. It exists in both graphical and command line modes. After installation, if you just type fastqc without any parameters, it will bring up the graphical interface. For bioinformaticians, however, we like to work in command line mode as much as possible due to large number of files that we usually need to process in automated fashion. Today, we will use fastqc to inspect the
sequence quality of these 2 files you just downloaded from SRA.
In Unix environment, using this command on multiple sequences becomes easier because you can use wild card characters. For example, if you want to run fastqc and create reports that can be viewed, you can just type like this:
fastqc *.fastq
This means I am telling the fastqc tool to process any files in a given directory that ends with “.fastq” extension. I will show this below.
[12]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
fastqc *.fastq
Analysis complete for SRR12584454_1.fastq
Analysis complete for SRR12584454_2.fastq
Started analysis of SRR12584454_1.fastq
Approx 5% complete for SRR12584454_1.fastq
Approx 10% complete for SRR12584454_1.fastq
Approx 15% complete for SRR12584454_1.fastq
Approx 20% complete for SRR12584454_1.fastq
Approx 25% complete for SRR12584454_1.fastq
Approx 30% complete for SRR12584454_1.fastq
Approx 35% complete for SRR12584454_1.fastq
Approx 40% complete for SRR12584454_1.fastq
Approx 45% complete for SRR12584454_1.fastq
Approx 50% complete for SRR12584454_1.fastq
Approx 55% complete for SRR12584454_1.fastq
Approx 60% complete for SRR12584454_1.fastq
Approx 65% complete for SRR12584454_1.fastq
Approx 70% complete for SRR12584454_1.fastq
Approx 75% complete for SRR12584454_1.fastq
Approx 80% complete for SRR12584454_1.fastq
Approx 85% complete for SRR12584454_1.fastq
Approx 90% complete for SRR12584454_1.fastq
Approx 95% complete for SRR12584454_1.fastq
Started analysis of SRR12584454_2.fastq
Approx 5% complete for SRR12584454_2.fastq
Approx 10% complete for SRR12584454_2.fastq
Approx 15% complete for SRR12584454_2.fastq
Approx 20% complete for SRR12584454_2.fastq
Approx 25% complete for SRR12584454_2.fastq
Approx 30% complete for SRR12584454_2.fastq
Approx 35% complete for SRR12584454_2.fastq
Approx 40% complete for SRR12584454_2.fastq
Approx 45% complete for SRR12584454_2.fastq
Approx 50% complete for SRR12584454_2.fastq
Approx 55% complete for SRR12584454_2.fastq
Approx 60% complete for SRR12584454_2.fastq
Approx 65% complete for SRR12584454_2.fastq
Approx 70% complete for SRR12584454_2.fastq
Approx 75% complete for SRR12584454_2.fastq
Approx 80% complete for SRR12584454_2.fastq
Approx 85% complete for SRR12584454_2.fastq
Approx 90% complete for SRR12584454_2.fastq
Approx 95% complete for SRR12584454_2.fastq
This is what you would see if you type the commands in your terminal. You can now inspect what files are being produced after the fastqc command was run.
[14]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
ls -la
total 169600
drwxr-xr-x 8 jsaw 982768932 256 Sep 9 09:41 .
drwxr-xr-x 6 jsaw 982768932 192 Sep 9 08:58 ..
-rw-r--r-- 1 jsaw 982768932 70168140 Sep 9 09:05 SRR12584454_1.fastq
-rw-r--r-- 1 jsaw 982768932 974550 Sep 9 09:41 SRR12584454_1_fastqc.html
-rw-r--r-- 1 jsaw 982768932 881128 Sep 9 09:41 SRR12584454_1_fastqc.zip
-rw-r--r-- 1 jsaw 982768932 70168140 Sep 9 09:05 SRR12584454_2.fastq
-rw-r--r-- 1 jsaw 982768932 973629 Sep 9 09:41 SRR12584454_2_fastqc.html
-rw-r--r-- 1 jsaw 982768932 867986 Sep 9 09:41 SRR12584454_2_fastqc.zip
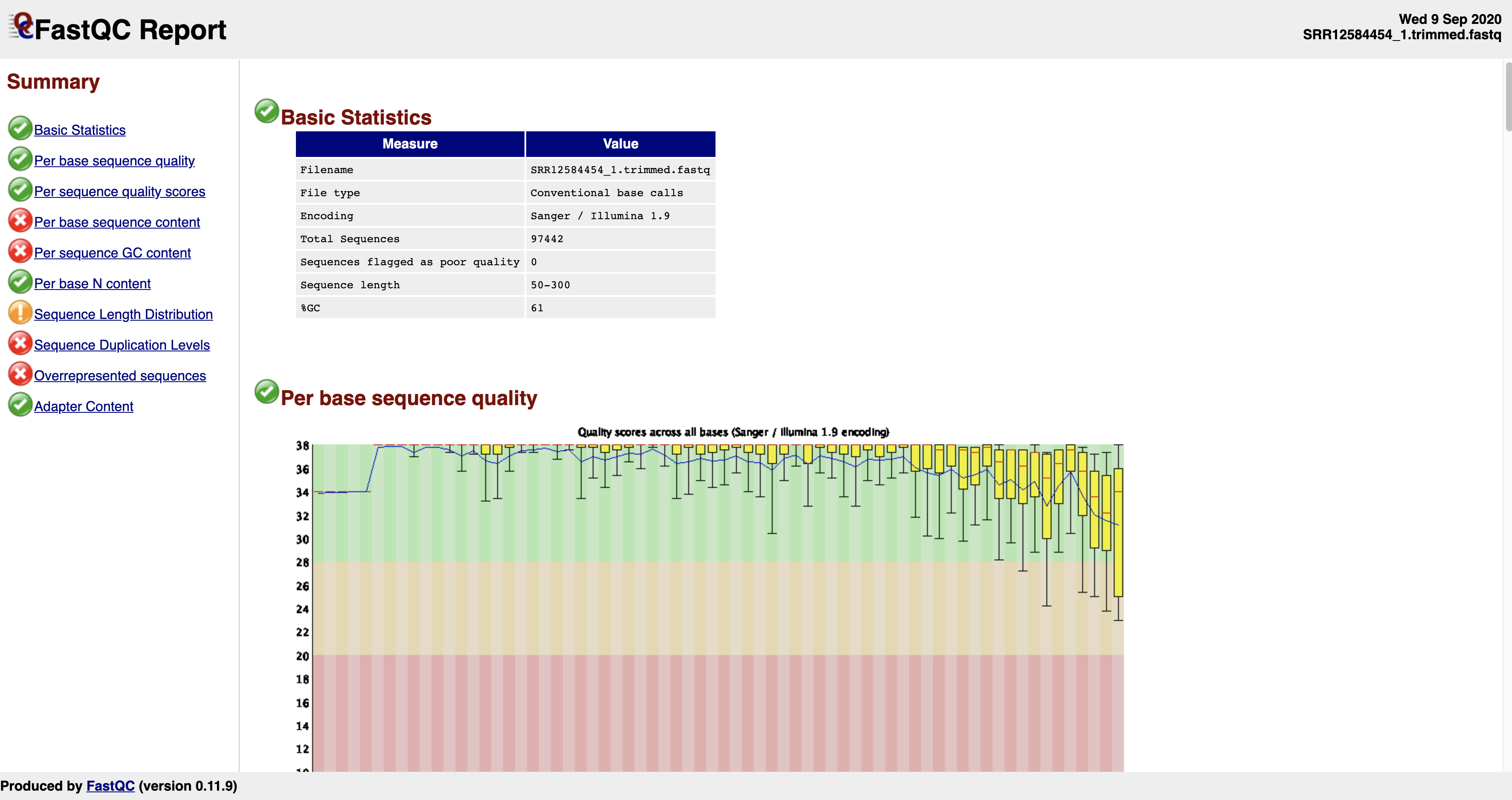
As you can see, the command produced an “html” and a “zip” file for each fastq. Now, open the html files using your web browser. You should see something similar to this:

This is just a screenshot of the very beginning of the report. As you scroll down, you will see more information. Basically, the check marks under “Summary” will tell you whether the sequences are good or bad. If you see multiple red crosses, that means the sequences are of poor or bad quality and shouldn’t be used right away until further processing is done. An example of a good and a bad sequencing run are shown here:
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/good_sequence_short_fastqc.html
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/bad_sequence_fastqc.html
A few things to note about the histograms in the first plot. You can see that each sequence is 301 basepair long and the histogram is basically trying to depict average sequence quality within a given window of sequence. You will notice that average sequence quality starts to drop as you go towards the end of the sequence. This is due to the nature of Illumina sequencing technology. It uses fluorescent molecules to record unique bases (A, C, G, T), and the fluorescence signal fades towards the later cycles of sequencing. This makes it difficult to confidently assign correct letters of DNA towards the end and the instrument records lower read qualities near the end.
Another thing you want to watch out for is the presence of “adapter” sequences (near the bottom of the report). See an example of bad sequence report. If the adapter sequences (which are artificial DNA constructs to facilitate sequencing) are left in the sequences for whatever reason, fastqc will detect it. In this bad sequence example, you will notice that the adapter content histogram starts to increase near the end of the sequences. If you see something like this, you will need to remove
the adapter sequences before the sequences can be used in genome assemblies or other analyses.
This is where the bbmap tool comes in.
A very useful website on how to read and interpret sequencing quality assessments: https://sequencing.qcfail.com/articles/?report=reader
3. BBTools¶
BBTools is a collection of tools written by scientists at the Joint Genome Institute (JGI) in Berkeley, CA. See here: https://jgi.doe.gov/data-and-tools/bbtools/
It contains a number of tools that can perform tasks such as interconversion of file formats, processing of raw sequencing files into usable ones, mapping of sequence reads to reference genomes, etc. The tool we will be using today is bbduk, which is meant for filtering and trimming of reads for adapter, contaminants, and quality using k-mers. Before you can actually use this tool, you need a reference file containing all known adapter sequences. bbduk can then look up for these
sequences to know if it can find these contaminants in your sequences.
First, download this adapter file here:
https://www.dropbox.com/s/f5mydteoupt8ugb/adapters.fa?dl=0
I suggest you put this “adapter.fa” file somewhere safe where there is no likelihood of it being deleted by accident.
Now, you can use bbduk to perform quality trimming and contaminant removal. To see what options are available with bbduk, type:
bbduk.sh -h
[15]:
%%bash
bbduk.sh -h
Written by Brian Bushnell
Last modified March 24, 2020
Description: Compares reads to the kmers in a reference dataset, optionally
allowing an edit distance. Splits the reads into two outputs - those that
match the reference, and those that don't. Can also trim (remove) the matching
parts of the reads rather than binning the reads.
Please read bbmap/docs/guides/BBDukGuide.txt for more information.
Usage: bbduk.sh in=<input file> out=<output file> ref=<contaminant files>
Input may be stdin or a fasta or fastq file, compressed or uncompressed.
If you pipe via stdin/stdout, please include the file type; e.g. for gzipped
fasta input, set in=stdin.fa.gz
Input parameters:
in=<file> Main input. in=stdin.fq will pipe from stdin.
in2=<file> Input for 2nd read of pairs in a different file.
ref=<file,file> Comma-delimited list of reference files.
In addition to filenames, you may also use the keywords:
adapters, artifacts, phix, lambda, pjet, mtst, kapa
literal=<seq,seq> Comma-delimited list of literal reference sequences.
touppercase=f (tuc) Change all bases upper-case.
interleaved=auto (int) t/f overrides interleaved autodetection.
qin=auto Input quality offset: 33 (Sanger), 64, or auto.
reads=-1 If positive, quit after processing X reads or pairs.
copyundefined=f (cu) Process non-AGCT IUPAC reference bases by making all
possible unambiguous copies. Intended for short motifs
or adapter barcodes, as time/memory use is exponential.
samplerate=1 Set lower to only process a fraction of input reads.
samref=<file> Optional reference fasta for processing sam files.
Output parameters:
out=<file> (outnonmatch) Write reads here that do not contain
kmers matching the database. 'out=stdout.fq' will pipe
to standard out.
out2=<file> (outnonmatch2) Use this to write 2nd read of pairs to a
different file.
outm=<file> (outmatch) Write reads here that fail filters. In default
kfilter mode, this means any read with a matching kmer.
In any mode, it also includes reads that fail filters such
as minlength, mingc, maxgc, entropy, etc. In other words,
it includes all reads that do not go to 'out'.
outm2=<file> (outmatch2) Use this to write 2nd read of pairs to a
different file.
outs=<file> (outsingle) Use this to write singleton reads whose mate
was trimmed shorter than minlen.
stats=<file> Write statistics about which contamininants were detected.
refstats=<file> Write statistics on a per-reference-file basis.
rpkm=<file> Write RPKM for each reference sequence (for RNA-seq).
dump=<file> Dump kmer tables to a file, in fasta format.
duk=<file> Write statistics in duk's format. *DEPRECATED*
nzo=t Only write statistics about ref sequences with nonzero hits.
overwrite=t (ow) Grant permission to overwrite files.
showspeed=t (ss) 'f' suppresses display of processing speed.
ziplevel=2 (zl) Compression level; 1 (min) through 9 (max).
fastawrap=70 Length of lines in fasta output.
qout=auto Output quality offset: 33 (Sanger), 64, or auto.
statscolumns=3 (cols) Number of columns for stats output, 3 or 5.
5 includes base counts.
rename=f Rename reads to indicate which sequences they matched.
refnames=f Use names of reference files rather than scaffold IDs.
trd=f Truncate read and ref names at the first whitespace.
ordered=f Set to true to output reads in same order as input.
maxbasesout=-1 If positive, quit after writing approximately this many
bases to out (outu/outnonmatch).
maxbasesoutm=-1 If positive, quit after writing approximately this many
bases to outm (outmatch).
json=f Print to screen in json format.
Histogram output parameters:
bhist=<file> Base composition histogram by position.
qhist=<file> Quality histogram by position.
qchist=<file> Count of bases with each quality value.
aqhist=<file> Histogram of average read quality.
bqhist=<file> Quality histogram designed for box plots.
lhist=<file> Read length histogram.
phist=<file> Polymer length histogram.
gchist=<file> Read GC content histogram.
enthist=<file> Read entropy histogram.
ihist=<file> Insert size histogram, for paired reads in mapped sam.
gcbins=100 Number gchist bins. Set to 'auto' to use read length.
maxhistlen=6000 Set an upper bound for histogram lengths; higher uses
more memory. The default is 6000 for some histograms
and 80000 for others.
Histograms for mapped sam/bam files only:
histbefore=t Calculate histograms from reads before processing.
ehist=<file> Errors-per-read histogram.
qahist=<file> Quality accuracy histogram of error rates versus quality
score.
indelhist=<file> Indel length histogram.
mhist=<file> Histogram of match, sub, del, and ins rates by position.
idhist=<file> Histogram of read count versus percent identity.
idbins=100 Number idhist bins. Set to 'auto' to use read length.
varfile=<file> Ignore substitution errors listed in this file when
calculating error rates. Can be generated with
CallVariants.
vcf=<file> Ignore substitution errors listed in this VCF file
when calculating error rates.
ignorevcfindels=t Also ignore indels listed in the VCF.
Processing parameters:
k=27 Kmer length used for finding contaminants. Contaminants
shorter than k will not be found. k must be at least 1.
rcomp=t Look for reverse-complements of kmers in addition to
forward kmers.
maskmiddle=t (mm) Treat the middle base of a kmer as a wildcard, to
increase sensitivity in the presence of errors.
minkmerhits=1 (mkh) Reads need at least this many matching kmers
to be considered as matching the reference.
minkmerfraction=0.0 (mkf) A reads needs at least this fraction of its total
kmers to hit a ref, in order to be considered a match.
If this and minkmerhits are set, the greater is used.
mincovfraction=0.0 (mcf) A reads needs at least this fraction of its total
bases to be covered by ref kmers to be considered a match.
If specified, mcf overrides mkh and mkf.
hammingdistance=0 (hdist) Maximum Hamming distance for ref kmers (subs only).
Memory use is proportional to (3*K)^hdist.
qhdist=0 Hamming distance for query kmers; impacts speed, not memory.
editdistance=0 (edist) Maximum edit distance from ref kmers (subs
and indels). Memory use is proportional to (8*K)^edist.
hammingdistance2=0 (hdist2) Sets hdist for short kmers, when using mink.
qhdist2=0 Sets qhdist for short kmers, when using mink.
editdistance2=0 (edist2) Sets edist for short kmers, when using mink.
forbidn=f (fn) Forbids matching of read kmers containing N.
By default, these will match a reference 'A' if
hdist>0 or edist>0, to increase sensitivity.
removeifeitherbad=t (rieb) Paired reads get sent to 'outmatch' if either is
match (or either is trimmed shorter than minlen).
Set to false to require both.
trimfailures=f Instead of discarding failed reads, trim them to 1bp.
This makes the statistics a bit odd.
findbestmatch=f (fbm) If multiple matches, associate read with sequence
sharing most kmers. Reduces speed.
skipr1=f Don't do kmer-based operations on read 1.
skipr2=f Don't do kmer-based operations on read 2.
ecco=f For overlapping paired reads only. Performs error-
correction with BBMerge prior to kmer operations.
recalibrate=f (recal) Recalibrate quality scores. Requires calibration
matrices generated by CalcTrueQuality.
sam=<file,file> If recalibration is desired, and matrices have not already
been generated, BBDuk will create them from the sam file.
amino=f Run in amino acid mode. Some features have not been
tested, but kmer-matching works fine. Maximum k is 12.
Speed and Memory parameters:
threads=auto (t) Set number of threads to use; default is number of
logical processors.
prealloc=f Preallocate memory in table. Allows faster table loading
and more efficient memory usage, for a large reference.
monitor=f Kill this process if it crashes. monitor=600,0.01 would
kill after 600 seconds under 1% usage.
minrskip=1 (mns) Force minimal skip interval when indexing reference
kmers. 1 means use all, 2 means use every other kmer, etc.
maxrskip=1 (mxs) Restrict maximal skip interval when indexing
reference kmers. Normally all are used for scaffolds<100kb,
but with longer scaffolds, up to maxrskip-1 are skipped.
rskip= Set both minrskip and maxrskip to the same value.
If not set, rskip will vary based on sequence length.
qskip=1 Skip query kmers to increase speed. 1 means use all.
speed=0 Ignore this fraction of kmer space (0-15 out of 16) in both
reads and reference. Increases speed and reduces memory.
Note: Do not use more than one of 'speed', 'qskip', and 'rskip'.
Trimming/Filtering/Masking parameters:
Note - if ktrim, kmask, and ksplit are unset, the default behavior is kfilter.
All kmer processing modes are mutually exclusive.
Reads only get sent to 'outm' purely based on kmer matches in kfilter mode.
ktrim=f Trim reads to remove bases matching reference kmers.
Values:
f (don't trim),
r (trim to the right),
l (trim to the left)
kmask= Replace bases matching ref kmers with another symbol.
Allows any non-whitespace character, and processes short
kmers on both ends if mink is set. 'kmask=lc' will
convert masked bases to lowercase.
maskfullycovered=f (mfc) Only mask bases that are fully covered by kmers.
ksplit=f For single-ended reads only. Reads will be split into
pairs around the kmer. If the kmer is at the end of the
read, it will be trimmed instead. Singletons will go to
out, and pairs will go to outm. Do not use ksplit with
other operations such as quality-trimming or filtering.
mink=0 Look for shorter kmers at read tips down to this length,
when k-trimming or masking. 0 means disabled. Enabling
this will disable maskmiddle.
qtrim=f Trim read ends to remove bases with quality below trimq.
Performed AFTER looking for kmers. Values:
rl (trim both ends),
f (neither end),
r (right end only),
l (left end only),
w (sliding window).
trimq=6 Regions with average quality BELOW this will be trimmed,
if qtrim is set to something other than f. Can be a
floating-point number like 7.3.
trimclip=f Trim soft-clipped bases from sam files.
minlength=10 (ml) Reads shorter than this after trimming will be
discarded. Pairs will be discarded if both are shorter.
mlf=0 (minlengthfraction) Reads shorter than this fraction of
original length after trimming will be discarded.
maxlength= Reads longer than this after trimming will be discarded.
minavgquality=0 (maq) Reads with average quality (after trimming) below
this will be discarded.
maqb=0 If positive, calculate maq from this many initial bases.
minbasequality=0 (mbq) Reads with any base below this quality (after
trimming) will be discarded.
maxns=-1 If non-negative, reads with more Ns than this
(after trimming) will be discarded.
mcb=0 (minconsecutivebases) Discard reads without at least
this many consecutive called bases.
ottm=f (outputtrimmedtomatch) Output reads trimmed to shorter
than minlength to outm rather than discarding.
tp=0 (trimpad) Trim this much extra around matching kmers.
tbo=f (trimbyoverlap) Trim adapters based on where paired
reads overlap.
strictoverlap=t Adjust sensitivity for trimbyoverlap mode.
minoverlap=14 Require this many bases of overlap for detection.
mininsert=40 Require insert size of at least this for overlap.
Should be reduced to 16 for small RNA sequencing.
tpe=f (trimpairsevenly) When kmer right-trimming, trim both
reads to the minimum length of either.
forcetrimleft=0 (ftl) If positive, trim bases to the left of this position
(exclusive, 0-based).
forcetrimright=0 (ftr) If positive, trim bases to the right of this position
(exclusive, 0-based).
forcetrimright2=0 (ftr2) If positive, trim this many bases on the right end.
forcetrimmod=0 (ftm) If positive, right-trim length to be equal to zero,
modulo this number.
restrictleft=0 If positive, only look for kmer matches in the
leftmost X bases.
restrictright=0 If positive, only look for kmer matches in the
rightmost X bases.
mingc=0 Discard reads with GC content below this.
maxgc=1 Discard reads with GC content above this.
gcpairs=t Use average GC of paired reads.
Also affects gchist.
tossjunk=f Discard reads with invalid characters as bases.
swift=f Trim Swift sequences: Trailing C/T/N R1, leading G/A/N R2.
Header-parsing parameters - these require Illumina headers:
chastityfilter=f (cf) Discard reads with id containing ' 1:Y:' or ' 2:Y:'.
barcodefilter=f Remove reads with unexpected barcodes if barcodes is set,
or barcodes containing 'N' otherwise. A barcode must be
the last part of the read header. Values:
t: Remove reads with bad barcodes.
f: Ignore barcodes.
crash: Crash upon encountering bad barcodes.
barcodes= Comma-delimited list of barcodes or files of barcodes.
xmin=-1 If positive, discard reads with a lesser X coordinate.
ymin=-1 If positive, discard reads with a lesser Y coordinate.
xmax=-1 If positive, discard reads with a greater X coordinate.
ymax=-1 If positive, discard reads with a greater Y coordinate.
Polymer trimming:
trimpolya=0 If greater than 0, trim poly-A or poly-T tails of
at least this length on either end of reads.
trimpolygleft=0 If greater than 0, trim poly-G prefixes of at least this
length on the left end of reads. Does not trim poly-C.
trimpolygright=0 If greater than 0, trim poly-G tails of at least this
length on the right end of reads. Does not trim poly-C.
trimpolyg=0 This sets both left and right at once.
filterpolyg=0 If greater than 0, remove reads with a poly-G prefix of
at least this length (on the left).
Note: there are also equivalent poly-C flags.
Polymer tracking:
pratio=base,base 'pratio=G,C' will print the ratio of G to C polymers.
plen=20 Length of homopolymers to count.
Entropy/Complexity parameters:
entropy=-1 Set between 0 and 1 to filter reads with entropy below
that value. Higher is more stringent.
entropywindow=50 Calculate entropy using a sliding window of this length.
entropyk=5 Calculate entropy using kmers of this length.
minbasefrequency=0 Discard reads with a minimum base frequency below this.
entropytrim=f Values:
f: (false) Do not entropy-trim.
r: (right) Trim low entropy on the right end only.
l: (left) Trim low entropy on the left end only.
rl: (both) Trim low entropy on both ends.
entropymask=f Values:
f: (filter) Discard low-entropy sequences.
t: (true) Mask low-entropy parts of sequences with N.
lc: Change low-entropy parts of sequences to lowercase.
entropymark=f Mark each base with its entropy value. This is on a scale
of 0-41 and is reported as quality scores, so the output
should be fastq or fasta+qual.
NOTE: If set, entropytrim overrides entropymask.
Cardinality estimation:
cardinality=f (loglog) Count unique kmers using the LogLog algorithm.
cardinalityout=f (loglogout) Count unique kmers in output reads.
loglogk=31 Use this kmer length for counting.
loglogbuckets=2048 Use this many buckets for counting.
khist=<file> Kmer frequency histogram; plots number of kmers versus
kmer depth. This is approximate.
khistout=<file> Kmer frequency histogram for output reads.
Java Parameters:
-Xmx This will set Java's memory usage, overriding autodetection.
-Xmx20g will
specify 20 gigs of RAM, and -Xmx200m will specify 200 megs.
The max is typically 85% of physical memory.
-eoom This flag will cause the process to exit if an
out-of-memory exception occurs. Requires Java 8u92+.
-da Disable assertions.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter any problems.
As you can see, the tool was written by Brian Bushnell and there are a lot of options you can specify with this tool. It can be pretty overwhelming to read and understand each of the parameter available for you to use. So I am giving you an example command with parameters I use for routing processing of raw sequence files.
bbduk.sh ktrim=r ordered minlen=50 mink=11 tbo rcomp=f k=21 ow=t ftm=5 zl=4 \
qtrim=rl trimq=20 \
in1=SRR12584454_1.fastq \
in2=SRR12584454_2.fastq \
ref=adapters.fa \
out1=SRR12584454_1.trimmed.fastq \
out2=SRR12584454_2.trimmed.fastq
Here you will notice a few things that are important in this example. First, I am telling bbduk to only keep sequences longer than 50 bases and quality higher than 20. And I am specifying the adapter file in ref=adapters.fa flag. in1 and in2 are where you specify input fastq files and out1 and out2 are where you specify output files, which should be named differently so that the program doesn’t overwrite the original files. I will show what it looks like when you type
this command.
[16]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
bbduk.sh ktrim=r ordered minlen=50 mink=11 tbo rcomp=f k=21 ow=t ftm=5 zl=4 \
qtrim=rl trimq=20 \
in1=SRR12584454_1.fastq \
in2=SRR12584454_2.fastq \
ref=adapters.fa \
out1=SRR12584454_1.trimmed.fastq \
out2=SRR12584454_2.trimmed.fastq
/Users/jsaw/miniconda3/opt/bbmap-38.86-0//calcmem.sh: line 75: [: -v: unary operator expected
Max memory cannot be determined. Attempting to use 1400 MB.
If this fails, please add the -Xmx flag (e.g. -Xmx24g) to your command,
or run this program qsubbed or from a qlogin session on Genepool, or set ulimit to an appropriate value.
java -ea -Xmx1400m -Xms1400m -cp /Users/jsaw/miniconda3/opt/bbmap-38.86-0/current/ jgi.BBDuk ktrim=r ordered minlen=50 mink=11 tbo rcomp=f k=21 ow=t ftm=5 zl=4 qtrim=rl trimq=20 in1=SRR12584454_1.fastq in2=SRR12584454_2.fastq ref=adapters.fa out1=SRR12584454_1.trimmed.fastq out2=SRR12584454_2.trimmed.fastq
Executing jgi.BBDuk [ktrim=r, ordered, minlen=50, mink=11, tbo, rcomp=f, k=21, ow=t, ftm=5, zl=4, qtrim=rl, trimq=20, in1=SRR12584454_1.fastq, in2=SRR12584454_2.fastq, ref=adapters.fa, out1=SRR12584454_1.trimmed.fastq, out2=SRR12584454_2.trimmed.fastq]
Version 38.86
Set ORDERED to true
maskMiddle was disabled because useShortKmers=true
0.038 seconds.
Initial:
Memory: max=1468m, total=1468m, free=1439m, used=29m
Added 3225 kmers; time: 0.038 seconds.
Memory: max=1468m, total=1468m, free=1433m, used=35m
Input is being processed as paired
Started output streams: 0.034 seconds.
Processing time: 6.863 seconds.
Input: 207684 reads 62512884 bases.
QTrimmed: 155680 reads (74.96%) 14257391 bases (22.81%)
FTrimmed: 207684 reads (100.00%) 207684 bases (0.33%)
KTrimmed: 128 reads (0.06%) 25083 bases (0.04%)
Trimmed by overlap: 167802 reads (80.80%) 1345557 bases (2.15%)
Total Removed: 12800 reads (6.16%) 15835715 bases (25.33%)
Result: 194884 reads (93.84%) 46677169 bases (74.67%)
Time: 6.937 seconds.
Reads Processed: 207k 29.94k reads/sec
Bases Processed: 62512k 9.01m bases/sec
When the tool finishes this task (which is pretty fast), you will see that it found 207,684 reads (if you combine the 2 fastq files) and removed 12,800 reads that do not meet the criteria I have set. And it retained 93.84% of the reads. The whole process only took less than 7 seconds. Now, if you look into the folder, you will notice 2 new files being produced that end with “.trimmed.fastq”.
[18]:
%%bash
cd ~/workspace/MicrobialGenomicsLab/exercises/
ls -lah
total 238M
drwxr-xr-x 11 jsaw 982768932 352 Sep 9 10:29 .
drwxr-xr-x 6 jsaw 982768932 192 Sep 9 08:58 ..
-rw-r--r-- 1 jsaw 982768932 67M Sep 9 09:05 SRR12584454_1.fastq
-rw-r--r-- 1 jsaw 982768932 53M Sep 9 10:29 SRR12584454_1.trimmed.fastq
-rw-r--r-- 1 jsaw 982768932 952K Sep 9 09:41 SRR12584454_1_fastqc.html
-rw-r--r-- 1 jsaw 982768932 861K Sep 9 09:41 SRR12584454_1_fastqc.zip
-rw-r--r-- 1 jsaw 982768932 67M Sep 9 09:05 SRR12584454_2.fastq
-rw-r--r-- 1 jsaw 982768932 44M Sep 9 10:29 SRR12584454_2.trimmed.fastq
-rw-r--r-- 1 jsaw 982768932 951K Sep 9 09:41 SRR12584454_2_fastqc.html
-rw-r--r-- 1 jsaw 982768932 848K Sep 9 09:41 SRR12584454_2_fastqc.zip
-rwxr-x--- 1 jsaw 982768932 14K Sep 9 10:28 adapters.fa
The next step after this is to check if sequence qualities have improved after you have run bbduk tool. To do that, you run fastqc tool again but on the trimmed fastq files. Type:
fastqc *.trimmed.fastq
And inspect the sequence qualities by opening the html files produced by fastqc. Noticed any differences? You can open the fastqc report of the original file in another tab to see how different things are. You should see something like this:
As you might notice, the histogram denoting sequence quality have drastically improved (no more columns dipping into the red zone). Now, the sequences are good to be used in downstream processes.
4. Preparations for next week’s lab¶
We will be doing genome and metagenome assemblies next week. For that, you will download a few publicly available datasets and also familiarize yourself with using ssh and rsync tools to remotely connect to and transfer files to other computers. You can look at this page for some examples of how to use ssh.
https://phoenixnap.com/kb/linux-ssh-commands
Another tool that will be crucial for you to use next week will be this tool known as rsync. It is a command line tool that can be used to transfer files between two remote computers or even for syncing directories inside the same computer. You can see some examples of how to use rsync here:
https://www.tecmint.com/rsync-local-remote-file-synchronization-commands/
and here:
https://phoenixnap.com/kb/rsync-command-linux-examples
Try to play around with a few examples shown in these pages to become familar with how rsync works.
Download data for next week¶
We will be using publicly available sequence data from NCBI SRA and see if you can replicate their results. The first one to download is here:
https://www.ncbi.nlm.nih.gov/sra/SRX9094324
These are Illumina MiSeq sequences they have used to reconstruct the genome of a Salmonella enteria strain. This is a cultured isolate and represents a single organism. Use fasterq-dump tool to download this data. I recommend you put it in data folder on your computer. These are fairly large files (each about 450 MB). In order to save space, you can compress them after they are downloaded. You can type like this:
gzip *.fastq
And this will compress your text files into much smaller zipped files with extension “*.gz”. The file sizes are much smaller after being compressed (about 110-130 MB).
For metagenome assemblies, we will be downloading this file:
https://www.ncbi.nlm.nih.gov/sra/SRX4741377
Make sure you use fasterq-dump to download it. Again, I recommend you put it in data folder on your computer and also use gzip to compress the fastq files as these are very large files (each about 4.5 GB, a total of 9 GB). These compressed files can be used directly by both fastqc and bbduk.
Now you are ready for next week’s lab.